Transformation Lab
A Plasmid Discovery
Labratory 6, AP Biology
Abstract. In the Transformation Lab designed by the Carolina Biological Supply Co., we took extracted DNA and inserted them into E. Coli bacterial cells through the transformation process (Carolina Biological Supply Co. 2014). We inserted genes for ampicillin resistance and green fluorescence, two genes not normally found in the bacteria. The genes were successfully inserted, yielding a ampicillin resistant, fluorescent E. Coli strand.
Introduction
The primary purpose of this experiment was to determine the different ways bacteria exchanges DNA, and learn about transformation efficiency. By studying the behaviors and mannerisms of the bacteria, we were able to observe these organisms on a deeper level. E. coli, a rod shaped bacterium normally found in the intestine, tends to shift depending on temperature and is more likely to grow when exposed to a non-sterile environment. Antibiotic techniques must be used to protect the E. Coli as well as yield results that embody the different environments each petri dish replicates. We inserted DNA into E. Coli to determine if there was any growth, and the results are described in the following experiment (Carolina Biological Supply Co., 2014)
Methods
On the first day we poured our agar plates. We first used a bottle of LB agar to pour 18 LB plates. We used another bottle to pour 2 LB plates and 16 LB/amp plates. We let the agar cool and then poured 2 more LB plates. We added a 4 mL vial of ampicillin to the remaining bottle of agar. We poured 16 LB/amp plates. We used the last bottle to pour the 16 LB/kanamycin plates. Once the agar cooled, we added a 4 mL vial of kanamycin to the 400 mL bottle. We poured 16 LB/kanamycin plates.
On the second day we began by labelling the starter plates with E. Coli and the date. We then sterilized the end of the inoculating tube with a flame from a lighter, making sure not to set it down afterwards. We uncapped the vial cap of the slant culture of E. Coli and passed the mouth of the vial through the flame to sterilize it. We then dragged the inoculating loop across an area of E. coli culture where there was obvious growth. Immediately after, we removed the loop, flamed the mouth of the vial, recapped the vial and set it to the side. We then used the inoculating tube to streak E. coli onto the agar plates, using proper technique, quickly replacing the lid. Immediately after we reflamed the loop to sterilize it. We did the same thing for each plate. We incubated the starter plates upside down in the incubator for 12-20 hours at 37 degrees Celsius.
Using sterile technique, on the third day 2 15 mL sterile tubes, one with +plasmid and one with -plasmid. We used a sterile transfer pipette to add 150 µL of ice-cold calcium chloride to both tubes, then we placed each on ice. We used a sterile inoculating loop to transfer isolated colonies of E. coli from the starter plate to the +plasmid tube. Immediately after, we suspended the cells by pipetting the liquid in and out with a sterile transfer pipette, then returned the +plasmid tube to the ice. We transferred a mass of cells to the -plasmid tube and suspended the liquid like above, then returned the tube to the ice so that both the +plasmid and the -plasmid tubes are on ice. Using a sterile disposable inoculating loop we added one loopful of plasmid DNA to the +plasmid tube. If we saw the clear, almost bubbly film across the loop the DNA was equal to 10 µL. We immersed the loopful of plasmid DNA into the cell suspension to mix the DNA with the cells. We then incubated both tubes in the ice for 15 minutes. After the incubation, we heat shocked both tubes by transferring them from the ice directly into a 42 degrees Celsius water bath for 90 seconds, agitating the tubes during the water bath. Immediately after the 9- second water bath we returned the tubes to the ice for another minute. Using a sterile transfer pipette we we added 250 µL of Luria broth to each tube, gently agitating the tubes so that it mixes well. We then let the tubes sit in room temperature for 15 minutes to recover.
We then placed the plates lid-side down to prepare for the glass bead spreading technique. We opened the plate and added 4-6 beads onto the lid, closing the plate and flipping it over immediately after. With the beads now resting on the agar, we used a sterile transfer pipette to add 100 µL of cells from the +plasmid and -plasmid tubes to each of the appropriate plates. Moving the glass beads in a back and forth motion, we spread the cells around the agar, letting the plates sit afterwards to allow time for the agar to absorb the cells. We then removed the glass beads by quickly dumping them into a bucket filled with bleach water. Finally, we stacked our plates and incubated them (Figures 5 & 6, Post-Experiment Figures 2-4)(Carolina Biological Supply Co., 2014).
Results
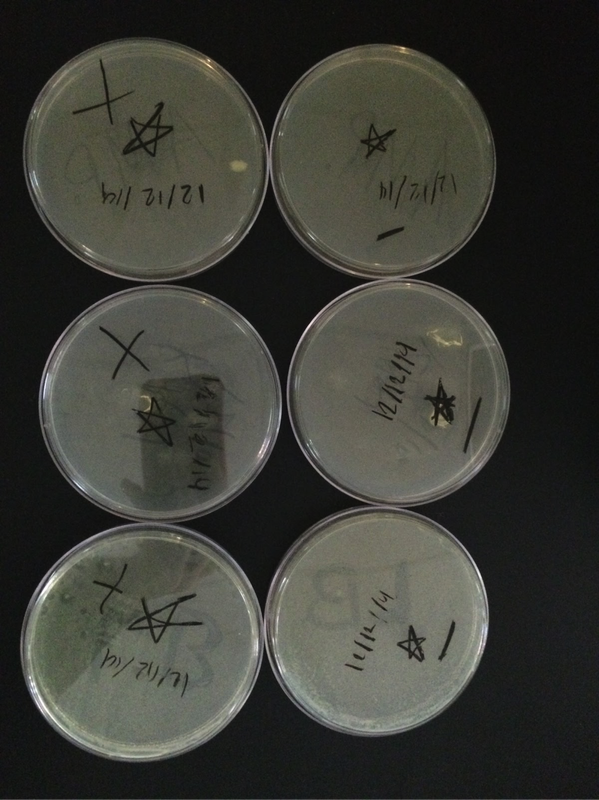
Once experimentation was completed, we viewed our results (Figures 1-4). Our ampicillin resistant (amp +) genes were viewed on the ampicillin plate in the form of a lawn, giving a confirmed positive result for that strain. We viewed no results in the amp- plate, as anticipated. In the normal LB plates, we viewed an expected, especially fluorescent, colony on the outskirts of the plate. Unfortunately, our other LB plate failed to yield any growth of anything. We gained no results on our kan(- and +) plates, as anticipated (Carroll, Chowdhury, Fox, Rodriguez, Thomas, 2014).
Discussion:
After examination, our results proved to be conclusive to an extent. All expected outcomes were achieved except for in our LB plate, which contained regular E.coli, there were signs of no growth (Carroll, Chowdhury, Fox, Rodriguez, Thomas, 2014). That could have been due to the lack of precision while streaking, or perhaps there was no bacteria on our streaker to begin with. There are a lot of factors that could have attributed to the inconclusive growth within the regular LB plate, had it gone correctly there should have been no complications with the growth and the E.coli would have grown normally given that it was ideal growing conditions for such bacteria.
Although that one plate was inconclusive our other plates did go as should. We saw the correct results within the ampR plate, signaling to us that the transformation went as planned. We cannot speak on behalf of the efficiency of the ampR plate given the fact that we only had a small colony. It also had a slight glow to it, also showing that the kan-resistance that we incorporated was slightly embedded in the new bacteria’s DNA. It is very possible that the glow might have been stronger and or perhaps fully inserted had we looked at our plates after a more prolonged period of time, giving the transformation enough time to cycle through the new bacteria. Many of these mistakes could have been either slightly or completely avoided if this were to have been done in a professional setting and had equipment to match it. Rather, given our circumstances and materials provided for us, our results proved as best conclusive as we could’ve wanted and hoped for. Our overall outcome was achieved, as we got to see an actual transformation of bacteria occur, thus proving the theory of McCarthy and MacLeod (GNN, n.d.) (Carroll, Chowdhury, Fox, Rodriguez, Thomas, 2014).
Questions:
1)
A Plasmid Discovery
Labratory 6, AP Biology
Abstract. In the Transformation Lab designed by the Carolina Biological Supply Co., we took extracted DNA and inserted them into E. Coli bacterial cells through the transformation process (Carolina Biological Supply Co. 2014). We inserted genes for ampicillin resistance and green fluorescence, two genes not normally found in the bacteria. The genes were successfully inserted, yielding a ampicillin resistant, fluorescent E. Coli strand.
Introduction
The primary purpose of this experiment was to determine the different ways bacteria exchanges DNA, and learn about transformation efficiency. By studying the behaviors and mannerisms of the bacteria, we were able to observe these organisms on a deeper level. E. coli, a rod shaped bacterium normally found in the intestine, tends to shift depending on temperature and is more likely to grow when exposed to a non-sterile environment. Antibiotic techniques must be used to protect the E. Coli as well as yield results that embody the different environments each petri dish replicates. We inserted DNA into E. Coli to determine if there was any growth, and the results are described in the following experiment (Carolina Biological Supply Co., 2014)
Methods
On the first day we poured our agar plates. We first used a bottle of LB agar to pour 18 LB plates. We used another bottle to pour 2 LB plates and 16 LB/amp plates. We let the agar cool and then poured 2 more LB plates. We added a 4 mL vial of ampicillin to the remaining bottle of agar. We poured 16 LB/amp plates. We used the last bottle to pour the 16 LB/kanamycin plates. Once the agar cooled, we added a 4 mL vial of kanamycin to the 400 mL bottle. We poured 16 LB/kanamycin plates.
On the second day we began by labelling the starter plates with E. Coli and the date. We then sterilized the end of the inoculating tube with a flame from a lighter, making sure not to set it down afterwards. We uncapped the vial cap of the slant culture of E. Coli and passed the mouth of the vial through the flame to sterilize it. We then dragged the inoculating loop across an area of E. coli culture where there was obvious growth. Immediately after, we removed the loop, flamed the mouth of the vial, recapped the vial and set it to the side. We then used the inoculating tube to streak E. coli onto the agar plates, using proper technique, quickly replacing the lid. Immediately after we reflamed the loop to sterilize it. We did the same thing for each plate. We incubated the starter plates upside down in the incubator for 12-20 hours at 37 degrees Celsius.
Using sterile technique, on the third day 2 15 mL sterile tubes, one with +plasmid and one with -plasmid. We used a sterile transfer pipette to add 150 µL of ice-cold calcium chloride to both tubes, then we placed each on ice. We used a sterile inoculating loop to transfer isolated colonies of E. coli from the starter plate to the +plasmid tube. Immediately after, we suspended the cells by pipetting the liquid in and out with a sterile transfer pipette, then returned the +plasmid tube to the ice. We transferred a mass of cells to the -plasmid tube and suspended the liquid like above, then returned the tube to the ice so that both the +plasmid and the -plasmid tubes are on ice. Using a sterile disposable inoculating loop we added one loopful of plasmid DNA to the +plasmid tube. If we saw the clear, almost bubbly film across the loop the DNA was equal to 10 µL. We immersed the loopful of plasmid DNA into the cell suspension to mix the DNA with the cells. We then incubated both tubes in the ice for 15 minutes. After the incubation, we heat shocked both tubes by transferring them from the ice directly into a 42 degrees Celsius water bath for 90 seconds, agitating the tubes during the water bath. Immediately after the 9- second water bath we returned the tubes to the ice for another minute. Using a sterile transfer pipette we we added 250 µL of Luria broth to each tube, gently agitating the tubes so that it mixes well. We then let the tubes sit in room temperature for 15 minutes to recover.
We then placed the plates lid-side down to prepare for the glass bead spreading technique. We opened the plate and added 4-6 beads onto the lid, closing the plate and flipping it over immediately after. With the beads now resting on the agar, we used a sterile transfer pipette to add 100 µL of cells from the +plasmid and -plasmid tubes to each of the appropriate plates. Moving the glass beads in a back and forth motion, we spread the cells around the agar, letting the plates sit afterwards to allow time for the agar to absorb the cells. We then removed the glass beads by quickly dumping them into a bucket filled with bleach water. Finally, we stacked our plates and incubated them (Figures 5 & 6, Post-Experiment Figures 2-4)(Carolina Biological Supply Co., 2014).
Results
Once experimentation was completed, we viewed our results (Figures 1-4). Our ampicillin resistant (amp +) genes were viewed on the ampicillin plate in the form of a lawn, giving a confirmed positive result for that strain. We viewed no results in the amp- plate, as anticipated. In the normal LB plates, we viewed an expected, especially fluorescent, colony on the outskirts of the plate. Unfortunately, our other LB plate failed to yield any growth of anything. We gained no results on our kan(- and +) plates, as anticipated (Carroll, Chowdhury, Fox, Rodriguez, Thomas, 2014).
Discussion:
After examination, our results proved to be conclusive to an extent. All expected outcomes were achieved except for in our LB plate, which contained regular E.coli, there were signs of no growth (Carroll, Chowdhury, Fox, Rodriguez, Thomas, 2014). That could have been due to the lack of precision while streaking, or perhaps there was no bacteria on our streaker to begin with. There are a lot of factors that could have attributed to the inconclusive growth within the regular LB plate, had it gone correctly there should have been no complications with the growth and the E.coli would have grown normally given that it was ideal growing conditions for such bacteria.
Although that one plate was inconclusive our other plates did go as should. We saw the correct results within the ampR plate, signaling to us that the transformation went as planned. We cannot speak on behalf of the efficiency of the ampR plate given the fact that we only had a small colony. It also had a slight glow to it, also showing that the kan-resistance that we incorporated was slightly embedded in the new bacteria’s DNA. It is very possible that the glow might have been stronger and or perhaps fully inserted had we looked at our plates after a more prolonged period of time, giving the transformation enough time to cycle through the new bacteria. Many of these mistakes could have been either slightly or completely avoided if this were to have been done in a professional setting and had equipment to match it. Rather, given our circumstances and materials provided for us, our results proved as best conclusive as we could’ve wanted and hoped for. Our overall outcome was achieved, as we got to see an actual transformation of bacteria occur, thus proving the theory of McCarthy and MacLeod (GNN, n.d.) (Carroll, Chowdhury, Fox, Rodriguez, Thomas, 2014).
Questions:
1)
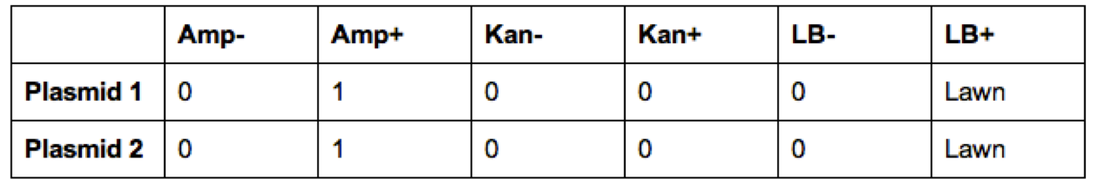
2) We expected 5 out of the 6 results. One of our normal agar plates turned out to not have any bacteria at all. This could be due to an improper streak technique, or even due to a low amount of bacteria on the actual streaker.
3)
- There may have been improper sterile technique used in the streaking phase, leading to absolutely no formation of bacteria. It could also be due to a lack of proper incubation.
- There seems to be an abnormally high amounts of bacteria growth in the ampicillin plate. It is as though someone either did not place the plasmids correctly, or did not use any (or not enough) ampicillin to get a good result.
- These results appear to have no errors.
4) a. 10 * 0.005 = 0.05 µL
b. 250 + 250 + 10 = volume in tube. 100 = spread amount. 100/510 = 0.196
c. The numbers are inaccurate, and the measurements taken were not with extremely accurate instruments, leaving multiple areas for error open.
5) 10/402 = 0.0249
6) x/25 = 0.01 x = 0.25
25*40 = 1000 mL
0.01*1000 = 10mL for amp solution, 990 mL for LB.
10x = 50 µg/mL
x = 5 mL
--------
1) When we inserted genes into our bacteria, we not only inserted the resistance genes, but also the genes to express fluorescence. These proteins take in blue light and reflect green light, thus drastically changing the phenotype of the bacteria.
2) The live bacteria was mixed in with the non-living bacteria, thus prompting the live bacteria to absorb the dead. In this process, the bacteria probably took in DNA from the dead cells, therefore transforming them into deadly cells.
3) Avery, Mccarthy, and Macleod extended on the previous knowledge from Griffith with experimentation on mice using the streptococcus pneumoniae which causes pneumonia in mammals. They then proceeded to inject the animals with a virulent S and nonvirulent R strand. The S strand evidently killed the mice and the R strand did not. Then when both S & R strands were killed via heat, neither strand killed the mice. They later mixed a live R strand with a dead S strand and found that it did in fact kill the mice, they then concluded that the nonvirulent strain had transformed into a virulent strain, through the passing of genetic material.
4) First I would isolate the gene needed to promote the reproduction of this protein. Then I would extract that gene, and insert bacteria into the pool of DNA, forcing transformation to occur. The bacteria would pick up the DNA and become useful to the patient.
5) Transformation(Take DNA from environment and integrate it into their own chromosome), conjugation(the transfer of DNA mediated by conjugal plasmids or conjugal transposons; requires cell to cell contact but can occur between distantly related bacteria or even bacteria and eukaryotic cells; can transfer long fragments of DNA), and transduction(phage doesn’t replicate immediately-dormant)
6) From the first and second assays, you could expect there to be more resistance in the second assay, due to the extended period of time the bacteria had to manipulate and take in the given plasmids. The first assay group would have had very little time to input the gene inside of them before they were sentenced to ampicillin, thus killing more (but not all) of the bacterial cells.
Conclusion
The purpose of this lab was to learn about the transformation efficiency of E. coli. The lab exemplifies ways that E.coli can gain antibiotic resistance. We now know that the colony with the ampicillin resistance had the greatest growth. We were able to artificially develop antibiotic resistance by inserting genes into the bacteria. This process is known as transformation. This can be beneficial to our final product as we learned about the nature of bacteria. Antibiotic resistance is detrimental in nature because the bacteria adapted to be able to fight off antibiotics. Antibiotic resistance is passed during replication. It is important to develop new antibiotics that the bacteria is not yet resistant to.
Citations
Carolina Transformation for AP Biology. (n.d.). Retrieved December 17, 2014, from https://echo.newtechnetwork.org/sites/default/files/new_uploads/20141211/_1418316053_Transformation Lab.pdf
Carroll, Chowdhury, Fox, Rodriguez, Thomas. (2014). Electrophoresis Student Input.
GNN - Genetics and Genomics Timeline. (n.d.). Retrieved December 17, 2014, from http://www.genomenewsnetwork.org/resources/timeline/1944_Avery.php
Photos and Charts: